Yeast RBPs¶
This tutorial compares RNAs UV-crosslinked to RNA-binding proteins (RBPs) Kre33 [Sharma-2017], Puf6 [Gerhardy-2021] and Nab3 [van.Nues-2017] in yeast Saccharomyces cerevisiae. The purpose is to show how similar experiments can be used as mutual controls for detecting unspecific background. Important is that the analysis is done with at least one protein known not to be directly involved in binding the same RNA molecules as the other proteins. Here, data for proteins proven to be involved in processing and assembly of pre-rRNA, Kre33 and Puf6, are set against data for the termination factor Nab3 to identify regions of the pre-rRNA that specifically associate to Kre33 or Puf6. More detailed analysis for these proteins is presented in the referred publications.
First,
create a work directory (say
Kre33Puf6/)
Then, move some files that have been shipped with the program:
copy the contents of this folder in the source ditribution coalispr/docs/_source/tutorials/Yeast/shared/ to
Kre33Puf6/.
In a terminal, change directory to the created work environment (from which all scripts and commands will be run):
cd /<path to>/Kre33Puf6/
Dataset¶
The yeast data we are using here have originally been aligned with Novoalign and analyzed with scripts from the pyCRAC suite [Webb-2014]. For evaluating the data with Coalispr we need bedgraph files. We can obtain these after aligning the data to the reference genome. To do this, the raw sequencing data is used and downloaded from the Gene Expression Omnibus (GEO) database with the relevant accession numbers retrieved from the literature:
CRAC-data |
Reference |
GEO acc. no. |
SRA table |
|---|---|---|---|
Kre33 |
|
||
Puf6 |
|
||
Nab3 |
|
Open the GEO Accession Display page for each of the experiments.
Enter the accession no. into the field
GEO accessionand pressGO.
For Kre33 and Puf6 all files are collected:
Access
SRA Run Selectorfrom bottom of the GEO accession display page that has been opened.On the SRA Run Selector webpage, click on the
Accession Listbutton in theTotalrow of theSelectpane.Save the file to the work directory and, because it will be combined with other data, add a prefix.
Save asKre33-SRR_Acc_List.txtandPuf6-SRR_Acc_List.txt.For detailed info, click the
Metadatabutton for a file describing the experiment.Save asKre33-SraRunTable.txtandPuf6-SraRunTable.txt
Collate the lists:
cat Kre33-SRR_Acc_List.txt Puf6-SRR_Acc_List.txt > SRR_Acc_List.txt
For Nab3 select the glucose tests on the SRA Run Selector webpage:
In the
Found # itemspane, selectSRR4024838,SRR4024839, andSRR4024840.In the
Selectpane, click on theSelectedbutton and then theAccession Listbutton.Save asNab3-SRR_Acc_List.txt.For experiment details, click the
Metadatabutton.Save asNab3-SraRunTable.txt.
Collate the lists:
cat Nab3-SRR_Acc_List.txt >> SRR_Acc_List.txt[1]
Download, extract fastq and compress the data (These steps create a directory structure (see Mouse miRNAs) in the working folder the scripts rely on).
sh 0_0-SRAaccess.shsh 0_1-SRAaccess.shsh 0_2-gzip-fastq.sh
Reference traces¶
Traces for gene-expression in cells grown under the same conditions support analysis. Coalispr can include these RNA-seq mRNA signals as reference traces. We would like to compare CRAC signals to mRNA signals. Although the next step can be included within the Nab3 downloads, often such data have to be downloaded from another GEO experiment, which we simulate here.
The mRNA reads for parental strain BY4740 linked to the Nab3 dataset will be used [van.Nues-2017]:
RNA-Seq |
Reference |
GEO acc. no. |
SRA table |
|---|---|---|---|
BY4741 |
|
Obtain the metadata as above, save as
BYRNASEQ-SraRunTable.txt.
Get the sequencing data (this step is optional; omit if reference traces are not needed) [5]:
prefetch SRR4024831sh 0_1_2-SRAaccess_ref.sh SRR4024831
We want to align the reference reads independently from the CRAC data [6] and for that change all SRR4024831 labels to refSRR4024831. First, SRR4024831-files are renamed and then the folder:
for i in 1 2; do mv SRR4024831/SRR4024831_$i.fastq SRR4024831/refSRR4024831_$i.fastq; done; mv SRR4024831 refSRR4024831;
Compress the uncollapsed reads.
sh 0_2-gzip-fastq.sh refSRR4024831
Alignment¶
In the other tutorials, sequence alignments were created with STAR; we will do that here as well. Before the data can be aligned to the yeast genome, we have to obtain reference files. The pyCRAC suite comes with reference fasta and gtf files for yeast, defining many non-coding RNAs [4]. More up-to-date versions have been used for the referenced papers and included here [7]. The GTF file should have been copied from the shared folder. Download an accompanying, sorted fasta genome:
wget -O - https://ftp.ensembl.org/pub/release-107/fasta/saccharomyces_cerevisiae/dna/Saccharomyces_cerevisiae.R64-1-1.dna_sm.toplevel.fa.gz | gunzip > saccer-R64.fasta
Then create the indices:
sh 1_1-star-indices.sh yeast saccer-R64.fasta Saccharomyces_cerevisiae.R64-1-1.75_1.2.gtf
The adapters have been removed from the reads deposited at GEO; still the 3’adapter (App_PE, see ‘Oligonucleotides for cDNA libraries’) is found in forward reads in reverse reads, maybe formed as primer-dimer, and some 5’ adapters. To remove these:
sh 2_0-flexbar-trim.sh
Make a dataset of collapsed reads:
sh 2_1-collapse-pre-trimmed-seqs.sh
The reads can now be aligned to the reference genome [8]. Because UV-crosslinking is an inefficient event, compared to normal RNA-IP, low numbers of RNAs are isolated that are specifically bound to the protein of interest. The covalently bonded RNA, after being released from this protein by enzymatic digestion with proteinase K [McKellar-2020], can still carry a crosslinked residue that will interfere with cDNA synthesis. These factors lead to reduced yields of specific sequences, enabling amplification of unspecific background. Isolation of cDNAs for specific RNAs is further affected when the RNA-binding protein of interest is low in abundance or its RNA-binding substrate has a strong, higher-order structure. A strategy to reduce the number of background reads in the analysis of crosslinked RNAs is to apply a high stringency at the mapping stage [9].
Due to UV-crosslinking, point-deletions and point-mutations are common in CRAC and CLIP-cDNAs. Therefore, such reads will be mapped while allowing at least an 1 nt mismatch;
Alignments and subsequent files will be stored in two directories within Kre33Puf6/ that will be created by the scripts, namely STAR-analysis1-yeast_collapsed/ and STAR-analysis1-yeast_uncollapsed/. The folder names contain the EXP parameter (i.e. ‘yeast’) which is the first argument for the mapping scripts. To align the collapsed reads with one mismatch [10] :
sh 3_1-run-starPE-collapsed-14mer.sh yeast 1
Do the same for the uncollapsed reads, run:
sh 4_1-run-starPE-uncollapsed-14mer.sh yeast 1
Remove the genome from shared memory (if that is used) when all mapping threads (set to 4 in the script) have completely finished:
sh 4_1_2-remove-genome-from-shared-memory.sh yeast
And create bedgraphs for both collapsed and uncollapsed data sets by [10]:
sh 3_2-run-make-bedgraphs.sh yeast 1sh 4_2-run-make-bedgraphs-uncollapsed.sh yeast 1
Note that the alignments are stored in their own subdirectories and that the filenames (Aligned.out.bam, Log.final.out etc.) are the same for each experiment. Therefore, Coalispr uses the folder names as a lead for retrieving bedgraph and bam files.
Align reference¶
If reference sequences have been downloaded (this was optional), these can be aligned as (un)collapsed reads and converted to bedgraphs after adding a third parameter to instruct the mapping scripts to process the reference data. Star will not see the readcounts for collapsed reads; if coverage is sufficient:
sh 2_1-collapse-pre-trimmed-seqs.sh refSRR4024831sh 3_1-run-starPE-collapsed-14mer.sh yeast 1 refSRR4024831sh 3_2-run-make-bedgraphs.sh yeast 1 refSRR4024831
For bedgraph values reflecting both coverage and numbers of mapped reads:
sh 4_1-run-starPE-uncollapsed-14mer.sh yeast 1 refSRR4024831sh 4_2-run-make-bedgraphs-uncollapsed.sh yeast 1 refSRR4024831
Coalispr¶
Described in the ‘How-to guides’ and like in the mouse tutorial we need to set up working conditions for the program. First a working environment is prepared and then the configuration files.
Work folder¶
Similar to the mouse tutorial we set up a workfolder for Coalispr inside Kre33Puf6/.
In a terminal change directory to
Kre33Puf6/and run:coalispr initGive ‘yeast’ as the EXP name for the session, in line with the alignments created above and confirm.
Choose the
currentfolder for setting up the Coalispr directory.
Output shows where the configuration files are:
Configuration files to edit are in:
'/<path to>/Kre33Puf6/Coalispr/config/constant_in'
The path '/<path to>/Kre33Puf6/Coalispr' will be set as 'SAVEIN' in 3_yeast.txt.
Configuration¶
Experiment file¶
As described in the How-to guides, for Coalispr a file describing the experiments, EXPFILE, has to be created. The fields (columns) required for the program are prepared in the XtraColumns.txt copied from Yeast-shared.
For an informative overview, not all columns or rows are collected and combined with XtraColumns.txt. Also, the csv format has to be changed to that of tsv. A separate script takes care of this:
copy python script
coalispr/coalispr/resources/share/cols_from_csv_to_tab.pyto the work directoryKre33Puf6/and run:python3 cols_from_csv_to_tab.py -f "Kre33-SraRunTable.txt,Puf6-SraRunTable.txt,Nab3-SraRunTable.txt,BYRNASEQ-SraRunTable.txt" -t2 -e XtraColumns.txt[12]- (
-fis input file,-t 2stands for “tutorial 2”, the one for yeast RNA binding proteins;-efor “expand with”)
The resulting file, Kre33Puf6_Exp.tsv, would be:
Run Description Short Category Group Method Fraction Experiment GEO_Accession (exp) Sample Name
SRR4305543 Kre33-data-I K33_a S K33 rip33 WCE SRX2199807 GSM2332452 GSM2332452
SRR4305544 Kre33-data-II K33_b S K33 rip33 WCE SRX2199808 GSM2332453 GSM2332453
SRR14570780 HTP-tagged Puf6 P6_a S P6 rip6 WCE SRX10913999 GSM5320150 GSM5320150
SRR14570781 HTP-tagged Puf6 P6_b S P6 rip6 WCE SRX10914000 GSM5320151 GSM5320151
SRR4024838 Nab3 Vari-X-link N3_a U N3 rip3 WCE SRX2016899 GSM2276892 GSM2276892
SRR4024839 Nab3 Megatron 1 N3_b U N3 rip3 WCE SRX2016900 GSM2276893 GSM2276893
SRR4024840 Nab3 Megatron 2 N3_c U N3 rip3 WCE SRX2016901 GSM2276894 GSM2276894
refSRR4024831 BY4741-RNA-Seq BY R NaN rnaseq WCE SRX2016892 GSM2276885 GSM2276885
Settings file¶
The next file to prepare is the Coalispr/config/constant_in/3_yeast.txt within Kre33Puf66/. This was copied from the 3_EXP.txt during coalispr init above, with some fields adapted to the current analysis. Edit this file:
scite Coalispr/config/constant_in/3_yeast.txt(by setting Language in the menu-bar to “Python” the active fields are highlighted compared to the comments)
Fields to be altered in the template (‘#’ indicates a comment):
EXP : “yeast” [13]
CONFNAM : “3_yeast.txt” [13]
EXPNAM : “Saccharomyces cerevisiae”
BINSTEP : 20 [14]
USEGAPS : BINSTEP [14]
MIRNAPKBUF : 1/4
SETBASE : “/<path to>/Kre33Puf6/”
MUTNO : “1” [11]
REFNAM : “refSRR4024831_”
REFS : REFNAM + TAG + “_” + MUTNO + “mismatch-” + EXP [15]
EXPFILNAM : “Kre33Puf6_Exp.tsv”
TOTAL : “rip3” [16]
RIP1 : “rip33” [17]
RIP2 : “rip6” [18]
EXPERIMENT : “Description”
- MUTGROUPS{
- “K33” : “Kre33”,“P6” : “Puf6”,“N3” : “Nab3”,}
- METHODS{
- TOTAL:”Nab3 crac”,RIP1:”Kre33 crac”,RIP2:”Puf6 crac”,}
UNSPECIFICS : [ “N3”, ]
MUTANTS : “”
LENGTHSNAM : “Saccharomyces_cerevisiae.R64-1-1.75_chromosome_lengths.txt”
GTFREFNAM : “Saccharomyces_cerevisiae.R64-1-1.75_1.2.gtf”
SAVEIN : Path(“/<path to>/Kre33Puf6/Coalispr”) [13]
EXPFILE : BASEDIR / EXPFILNAM
REFDIR : BASEDIR / SRCFLDR / REFS
GTFREF : BASEDIR / GTFREFNAM
Analysis¶
After preparing the configuration files, we can process the bedgraphs.
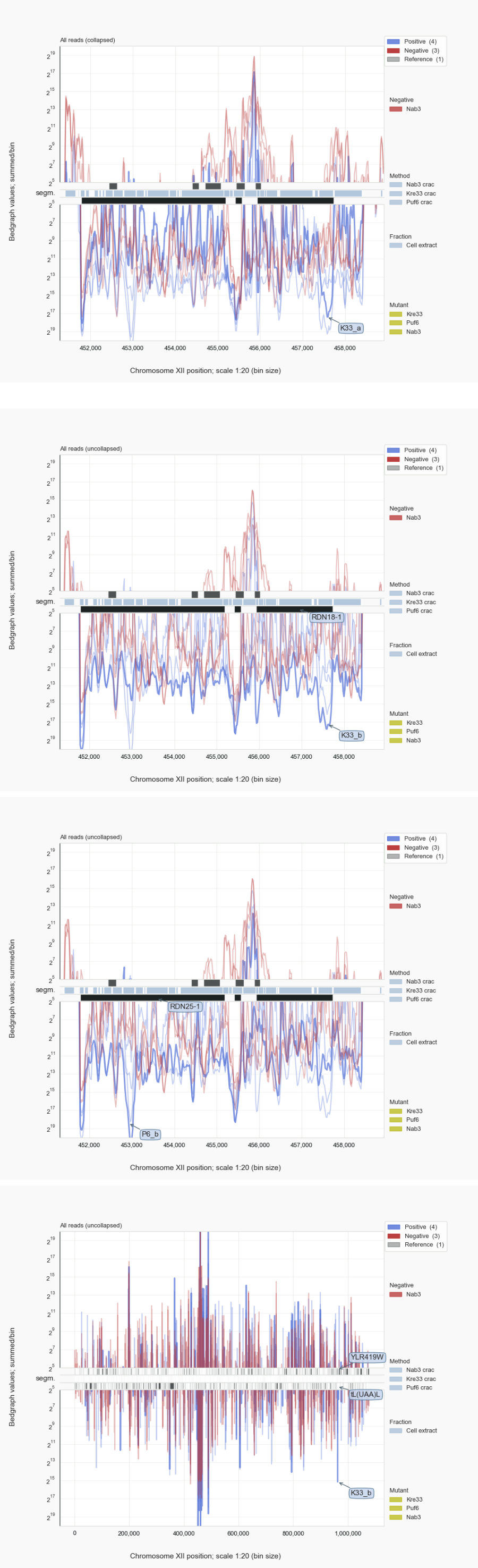
Figure 1. rRF cDNAs and reads¶
setexp¶
Begin with activating the new configuration:
cd /<path to>/Kre33Puf6/coalispr setexp -e yeast -p2choose option 2 and confirm.
storedata¶
Now the bedgraph data can be loaded into Pandas dataframes, saved as parquet files by Coalispr:
When reference bedgraphs are used, these can be stored using option -d2. First, do this for the uncollapsed reads (the default type, -t1):
coalispr storedata -d1coalispr storedata -d2
For collapsed reads, getting an idea of the different cDNAs crosslinked to the proteins, do:
coalispr storedata -d1 -t1coalispr storedata -d2 -t1
showgraphs¶
Analysis of bedgraph traces for all uncollapsed reads with
coalispr showgraphs -c XII -w2
or checking traces representing cDNAs with
coalispr showgraphs -c XII -w2 -t1
shows how specific rRFs for Kre33 derive from 18S regions in the rDNA and those for Puf6 from a 25S section (top panels in Fig. 1). Zooming out from the rDNA region (bottom panel in Fig. 1) shows how noisy the RNA-seq data from cross-linked samples can be (e.g. for Kre33 or Puf6 [22]). Therefore, monitoring single nt deletions (for CRAC) or mutations (for CLIP) that will be common for crosslinked uracil residues in crosslinked RNA fragments is an important measure. The pyCRAC software suite [Webb-2014] has been specifically developed for such in-depth analysis of RNA-seq data for crosslinked RNA. The referred publications for Kre33 [Sharma-2017], Puf6 [Gerhardy-2021] and Nab3 [van.Nues-2017] describe such an analysis. Here we aimed to illustrate usage of Coalispr and show how independent experiments can form mutual controls for particular observations, as mentioned in the essay “”.